
粉末衍射的结构解决方案
Endeavour是一个强大的软件晶体结构解决方案,从粉末和单晶衍射数据。基于十多年的经验,该软件能够或多或少地自行解决许多中小型结构。创新的概念与精心设计的用户界面相结合,使晶体结构的求解几乎成为一个常规的过程,尤其是对于无机物,也适用于许多有机化合物。
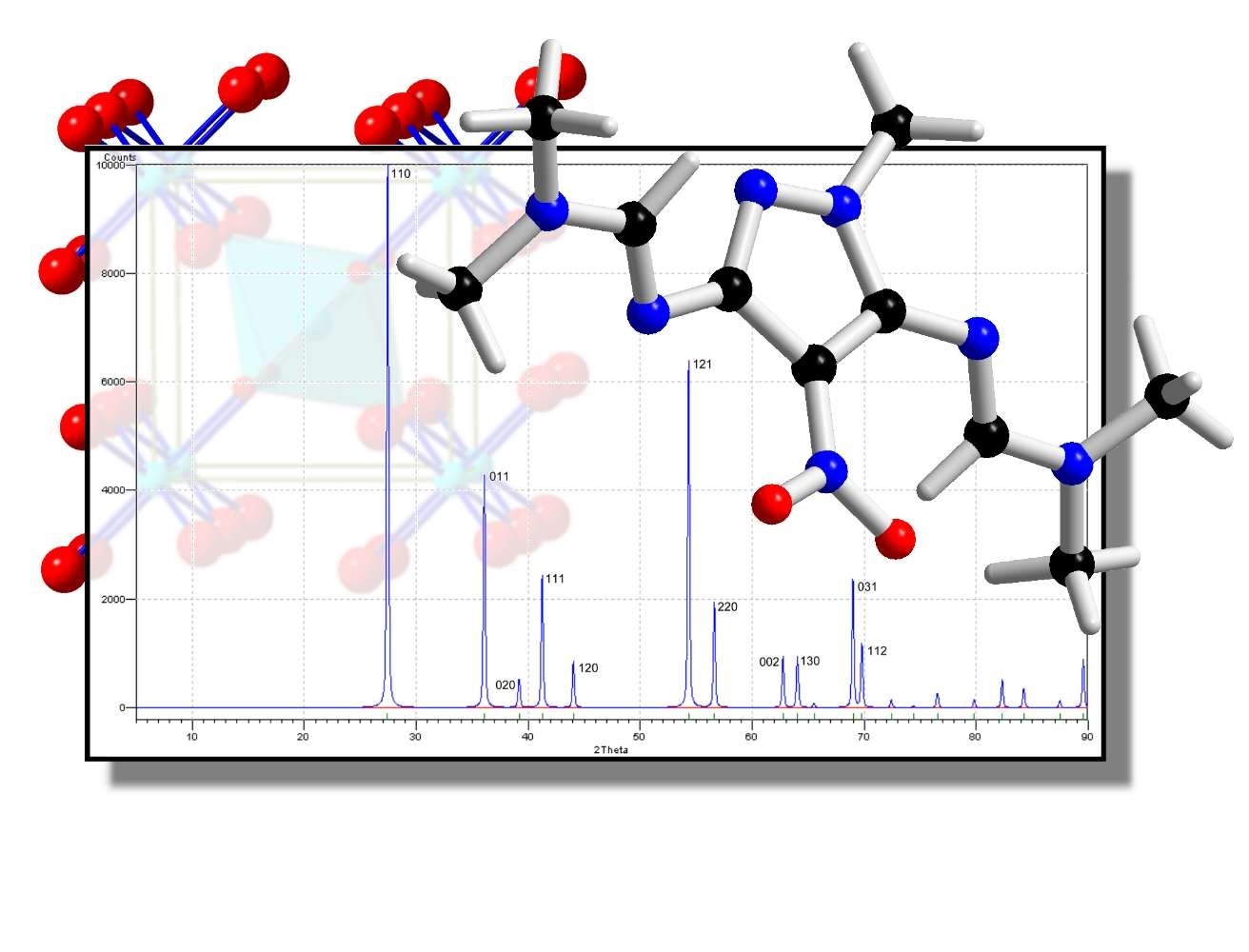
即使是没有经验的用户也可以通过几个步骤来准备和执行结构溶液计算:只需按照集成的“向导”输入所需的数据(单位单元参数、化学成分、衍射数据),然后让Endeavour完成剩下的工作。利用“直接空间”方法的一种特殊变种,即对计算和观测衍射数据之间的差异以及系统势能的组合全局优化,执行结构解决方案。由于势能的额外使用,与直接方法相比,该方法对低质量衍射数据的敏感性要低得多。
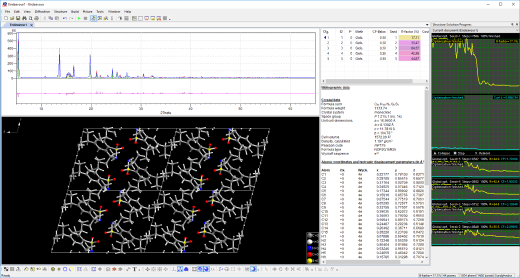
最新版本的1.8带有重新设计的用户界面,带来了一些改进和错误修复。
科学背景
《应用结晶学杂志:H.Putz,J.C.Schoen,M.Jansen》和《粉末衍射数据中“Ab Initio”结构溶液的组合方法》(J.Appl)上已经发表了关于Endeavour 的科学背景的详细资料。J. Appl. Cryst. (1999), 32,864-870。
目的
粉末或单晶衍射数据的晶体结构解决方案,使用计算和测量衍射图案和系统势能差异的综合全局优化(“Pareto优化”)。
功能
结构方案:
传统X射线实验室衍射仪以及同步加速器、中子或电子衍射数据的结构解决方案
需要输入:
单晶参数
衍射数据:粉末衍射数据(Bragg角处的综合强度(非剖面/阶跃扫描数据),从飞利浦、BrukerAXS、Stoe导入,或单晶衍射数据(I(hkl), |F(hkl)|或F2(hkl)),例如从SHELX文件导入。
单位细胞含量(组成或分子结构加上公式单位数)
“向导”,便于准备输入数据
自动调整所有结构解决方案计算参数
从实验和计算峰值列表中计算剖面图,而不仅仅是比较相关峰值的强度。这样做,R因子计算对2theta误差和错误的峰值分配就不那么敏感了。在早期版本的Endeavour中,这个问题经常导致错误的R因子值,并可能阻止整个晶体结构的解决。
根据生成的结构自动确定空间组
可选或自动检查/纠正2theta错误。
自动检查峰值相关性(可选,默认为激活)。
几个“高级设置”,例如柔性分子、跳跃宽度、峰值三角形、模拟退火参数等-可从结构解决方案向导中获得。
含有分子/刚性体的晶体结构现在可以在除P1以外的空间群中直接求解,即使它们的原子必须放置在特殊位置。
查看结构解决方案过程的中间步骤:进度(完成百分比)、R值和总体成本函数、相关计算和观察峰值列表、结构图和用户定义的并排排列的衍射图。
自动构建功能,以可视化每个中间步骤的晶体结构,例如单晶,有键或者无键,有无多面体、分子等。
报告视图,将Pareto优化结果显示为格式化文本,可以复制到剪贴板、保存或打印。
易于编辑、计算和拟合潜在参数(简单排斥,Lennard Jones)。
所有元素对组合(源自皮尔逊晶体数据)的最小原子间距离现已实现(不再需要手动调整简单排斥势的参数)。
对分子和原子晶体结构的支持。分子可以固定在特定的位置,并可以沿着参考原子旋转。
具有自动或交互式转换到更高空间组的对称查找器。
“结构解决方案”可通过能量最小化或衍射数据单独实现。手动选择分子内的可旋转键,以便在结构溶液计算过程中进行变化。
从各种文件格式导入三维分子结构,包括Diamond(*.dsf)、Cambridge CSD-FDAT(*.dat、.fdat、.csd)、MDL Molfile/ SDFile(例如,从ACD Chemsketch)(*.mol、.mdl、.sd)、Cerius2 CSSR(*.cssr、.dat)、Sybyl Mol/Mol2(*.mol、.mol2)和CIF(*.cif)文件。
还可以使用ACDChemSketch免费软件绘制分子草图并将其转换为3D,该软件可以从Endeavour 1.x CD-ROM安装。
结构可视化:
结构可视化“自动构建”功能(如分子、多面体)的设置现在可以自动调整,同时考虑到正在研究的化合物的“化学性质”。
手动输入或更改结构参数和书目数据。
从晶体结构数据库(无机晶体结构数据库、剑桥结构数据库、蛋白质数据库)、Pauling文件(无机材料数据库)、晶体学信息文件(CIF)、SHELX格式以及多种分子结构格式导入结构数据。
创建单晶、超级单细胞或任意范围的晶体晶格的内容。
通过显示所选原子类型之间距离分布的柱状图辅助的连接性讨论,以及自动计算距离范围。
完成选定原子周围的配位球。
生成分子或完成细胞边缘的分子片段(包装图)。
球棒、铁丝和空间填充模型。中央或平行投影、深度提示、立体显示。
具有用户定义光源和材料特性(OpenGL)的真实照片渲染模型。
旋转、移动和缩放:鼠标控制、键盘步进或数字。沿指定轴或朝向hkl平面的视图。
围绕选定原子的协调多面体,或手动构造,带有阴影、不透明或透明面。
显示热椭球体,以可视化各向异性位移参数。
标记原子和键。原子和键的颜色、样式和半径的变化,按类型或个别分配。
概述:
32位MS Windows应用程序,带有选项卡式多文档界面(MDI)、上下文相关菜单和工具栏。允许“同时”处理多个结构。
用户定义的结构图和衍射图以及多个文本数据并排排列。
多步骤撤销和重做功能。
计算或交互测量距离、角度和扭转角度,包括标准不确定性。
导出到晶体和分子结构格式、Peak和|F(hkl)|格式、Windows元文件、几种位图格式以及虚拟现实建模语言(VRML)。
在线更新(自动或手动)。
系统要求
带有MSWindows®XP、Vista、Windows 7、Windows 8/8.1或Windows 10操作系统的个人计算机(在“S”模式下不在Windows RT或Windows 10 S或Windows 10上运行)
Microsoft Internet Explorer 5.01或更高版本
64 MB内存
100 MB可用磁盘空间
图形分辨率为1024 x768像素,颜色为32768色(“高亮颜色”)。